研究内容

In-depth analysis reveals complex molecular aetiology in a cohort of idiopathic cerebral palsy. (Li N, et al., Brain, 2021)

Genetics of intellectual disability in consanguineous families. (Hu H, et al., Molecular Psychiatry, 2018)
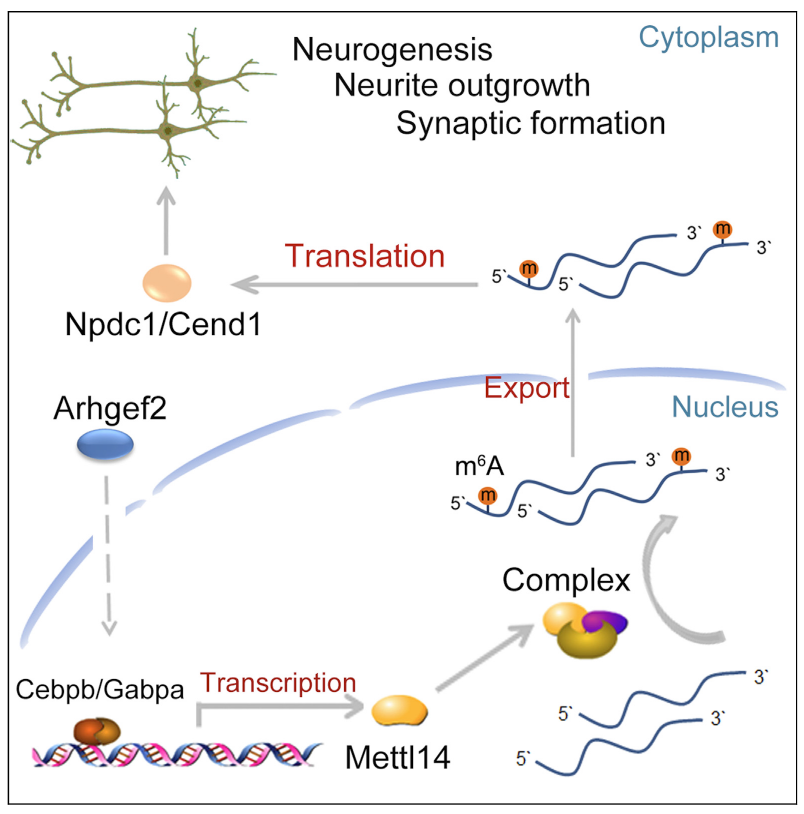
Arhgef2 regulates neural differentiation in the cerebral cortex through mRNA m(6)A-methylation of Npdc1 and Cend1. (Zhou P, et al., iScience, 2021)
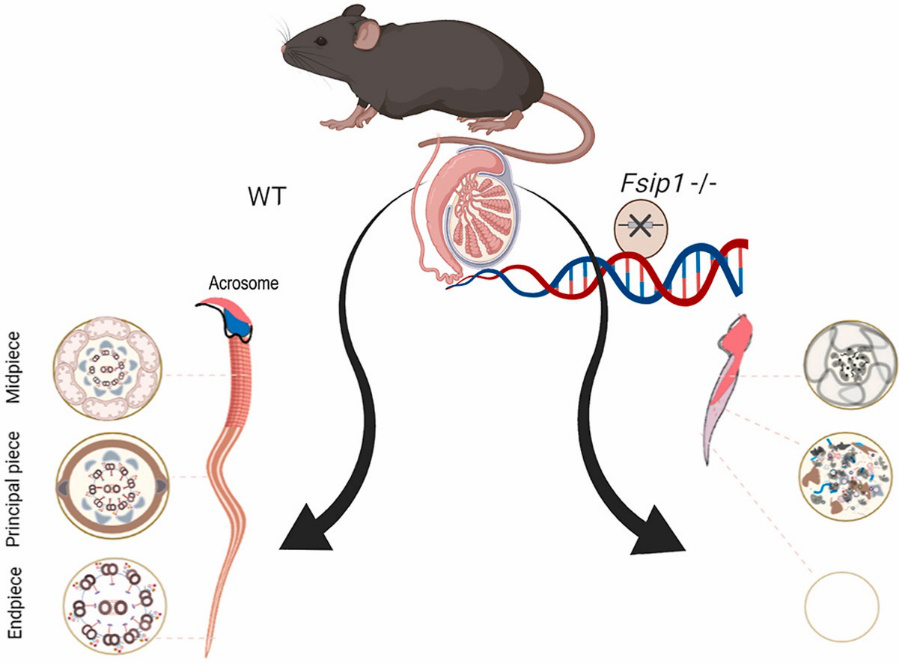
Bi-allelic mutation in Fsip1 impairs acrosome vesicle formation and attenuates flagellogenesis in mice. (Gamallat Y, et al., Redox Biol, 2021)
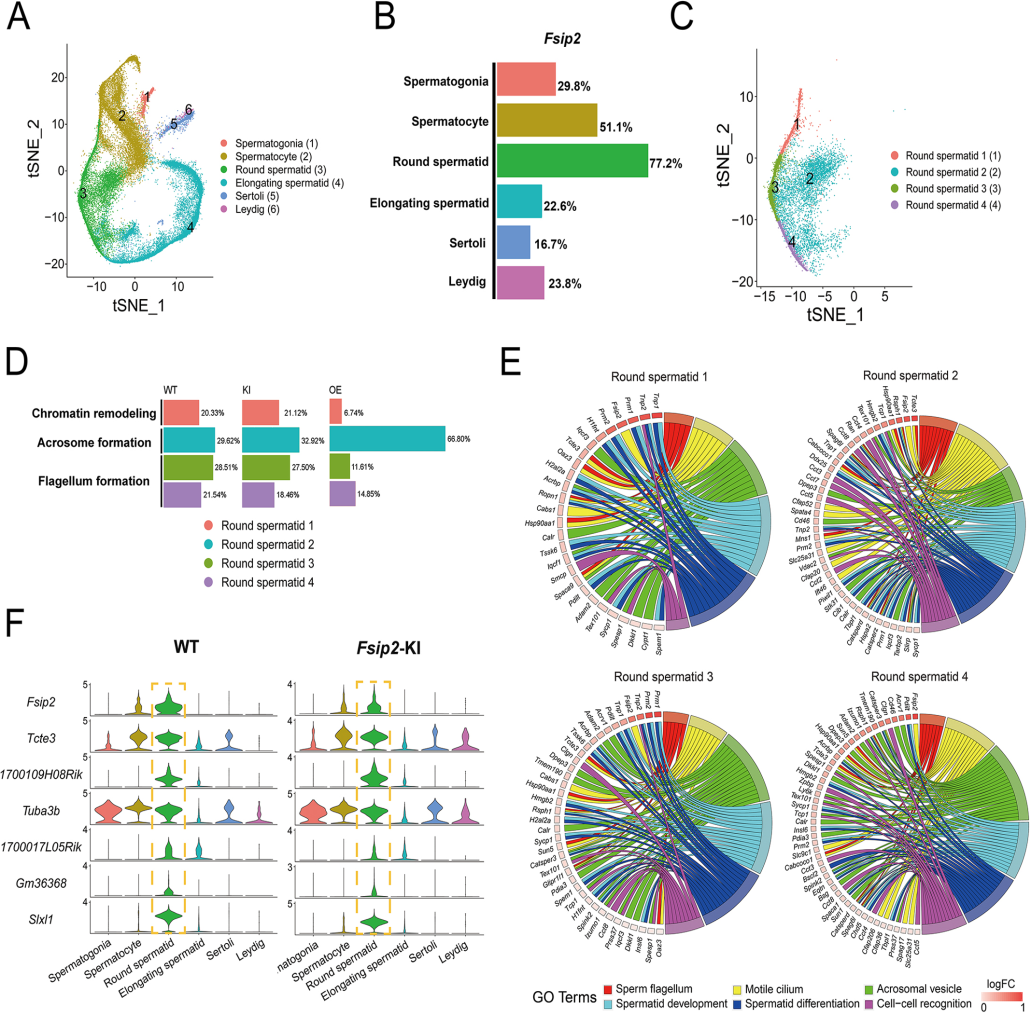
Hypomorphic and hypermorphic mouse models of Fsip2 indicate its dosage-dependent roles in sperm tail and acrosome formation. (Fang X, et al., Development, 2021)